Массообменные процессы
Лабораторная работа № 1
ОПРЕДЕЛЕНИЕ КОЭФФИЦИЕНТА МАССОПЕРЕДАЧИ В ПРОЦЕССЕ АБСОРБЦИИ
Цель работы: практическое изучение работы абсорбера и определение коэффициента массопередачи на основе полученных экспериментальных данных.
Абсорбцией называют процесс поглощения газа жидким поглотителем, в котором газ растворим в той или. иной степени.
На практике абсорбции подвергаются большей частью не отдельные газы, а газовые смеси, составные части которых могут поглощаться данным поглотителем в заметных количествах. Эти составные части называют абсорбируемыми компонентами, а непоглощаемые составные части - инертным газом.
В данной работе взяты: в качестве абсорбируемого компонента - аммиак, в качестве поглотителя (абсорбента) - вода. Воздух рассматривается как инертный газ, не растворимый в воде.
Описание лабораторной установки

Орошающая вода подается на верх колонны из водопроводной сети. Образовавшаяся в результате процесса абсорбции аммиачная вода слабой концентрации сбрасывается в слив, а очищенный воздух выбрасывается в атмосферу. Расходы воды и аммиака регулируются вентилями ВР1, ВР2 и замеряются соответственно ротаметрами 1 и 2. Уровнемер 5 позволяет следить за уровнем аммиачной воды в нижней части колонны. Содержание аммиака определяется отбором проб с последующим титрованием.
Порядок выполнения работы.
1. Открыть вентиль подачи воды и установить ее заданный расход.
2. Пустить воздуходувку и установить расход воздуха, соответствующий пленочному режиму работы колонны (визуально).
3. Открыть вентиль для подачи аммиака и установить его расход.
4. Через 10 минут отобрать пробы газа в точках 6 и 7 и пробу жидкости в точке 8, замерить температуру в точках 3 и 4.
5. Повторить отбор проб.
Рис. 1.1. Схема экспериментальной установки.
6. По окончании работы закрыть подачу аммиака, выключить газодувку и перекрыть воду.
Отбор проб и титрование
1. На содержание аммиака в воде: открыть пробоотборник 8 и налить в колбу 250 мл аммиачной воды; пипеткой отобрать в коническую колбу 10 мл раствора и разбавить дистиллированной водой; к полученному раствору добавить 3-4 капли фенолфталеина и титровать 0,1 н. раствором серной кислоты. Записать в табл. 1.1 количество израсходованной серной кислоты.
2. На содержание аммиака в газе: отобрать 100 мл газовой смеси с помощью шприца, медленно пропустить смесь через поглотительную склянку, в которой находится 10 мл 0,01 н. раствора H2SO4. Слить полученный раствор в коническую колбу, добавить 3-4 капли фенолфталеина и титровать 0,01 н. раствором NaOH. Записать в табл. 1.1 количество израсходованной щелочи.
Результаты анализа и замеров Таблица 1.1
|
Газ на входе в абсорбер |
Газ на выходе из абсорбера |
Вода на выходе из абсорбера |
Расход воды L, м3/ч |
Расход аммиака М, м3/ч |
Температура газа перед абсорбером t, oC |
Температура газа после абсорбера t1, oC |
|||||||
|
Количество 0,01 н. раствора H2SO4 в поглотительной склянке, мл |
Количество газа, пропущенного через поглотительную склянку, мл |
Количество 0,01 н. раствора NaOH, затраченного на титрование, мл |
Количество аммиака, поглощенного в склянке, мл |
Количество 0,01 н. раствора H2SO4 в поглотительной склянке, мл |
Количество газа, пропущенного через поглотительную склянку, мл |
Количество 0,01 н. раствора NaOH, затраченного на титрование, мл |
Количество аммиака, поглощенного в склянке, мл |
Количество воды, взятое на анализ, мл |
Количество 0,1 н. раствора H2SO4, затраченного на титрование, мл |
||||
Обработка опытных данных.
В пленочном режиме работы насадочной колонны поверхность контакта жидкости и газа F весьма близка к площади поверхности насадочных тел, и коэффициент массопередачи может быть определен из основного уравнения массопередачи [1]: M = Ky × DYср × F,
где F = H × f × s - поверхность фазового контакта, м2; H - высота слоя насадки, м; s - удельная поверхность насадки, м2/м3; f - площадь поперечного сечения колонны, м2; М - количество абсорбированного аммиака, кмоль/час; Кy - коэффициент массопередачи, отнесенный к концентрации газа, кмоль/(м2×ч); DYср - средняя движущая сила абсорбции в единицах концентрации газовой фазы, кмоль NH3/кмоль воздуха.
Расход воздуха и количество абсорбированного аммиака определяется материальным балансом абcорбции: M = L(Xк
- Хн) = Gв
(Yн - Yк),
где Gв —
расход воздуха, кмоль/час; L - расход воды, кмоль/час; Yн, Yк - концентрация аммиака в воздухе до и после колонны, кмоль NH3/кмоль воздуха; Xк, Хн - концентрация аммиака в воде на входе и выходе из колонны, кмоль NH3 / кмоль воды.
При небольших концентрациях раствора растворимость
аммиака в воде подчиняется закону Генри, и равновесие в системе следует уравнениям:
У* = 2,37 X при t = 10 oС; У* = 2,74 X при t = 20 °С.
Для этих условий движущая сила абсорбции может быть рассчитана как среднелогарифмическая величина: DYср = (DYб - DYм) / 2,3 lg (DYб /DYм),
где DYб = Yн - Yн* - движущая сила процесса внизу абсорбера; DYм = Yк
- Yк* - движущая сила процесса вверху абсорбера.
Рассчитанный коэффициент массопередачи Кy
соответствует скорости воздуха в свободном сечении колонны, определяемой по уравнению расхода:
W = Gв ×22,4 × (273 + t) / (273 × f ×3600).
Пример расчета концентраций
На титрование 10 мл 0,01н. раствора H2SO4, через который было пропущено 100 мл смеси воздуха с аммиаком, затрачено 6,8 мл 0,01н.
раствора NaOH.
Определить концентрацию аммиака в газовой смеси Ун.
1. Объем 0,01н. раствора H2SO4
прореагировавший с аммиаком смеси газа: H2SO4 = 10 - 6,8 = 3,2 мл
2. Количество аммиака в 100 мл смеси газа:
1 мл 0, 01 н. раствора H2SO4 реагирует с 0,224 мл NH3:
Х = 3,2 × 0,224 = 0,716 мл.
3. Содержание NH3
в 1 мл газа:
100 мл смеси - 0,716 мл NH3
1000 мл смеси - X мл NH3
Х = 7,16 мл NH3 / л смеси = 0,00716
л NH3/л смеси = 0,00716 / (1 - 0,00716) =
= 0,00721 кмоль NH3 / кмоль воздуха = Yн.
Аналогично определяем концентрацию Yк в газе на выходе.
На титрование 10 мл аммиака в воде затрачено 8 мл 0,1 н. раствора Н2SO4. Определить концентрацию аммиака в воде Xк, принимая Xн = 0.
1. В 1 л 0,1 н. раствора содержится 4,9 г Н2SO4
10 мл 0,1 н. раствора - 0,049 г Н2SO4
8 мл 0,1 н. раствора - X Н2SO4
X = 0,0392 г Н2SO4
2. Количество NH3 в 10 мл раствора NH4OH:
2 NH4OH + Н2SO4= (NH4)2SO4 + 2 Н2О
44,8 л NH3 - 98 г Н2SO4
Х л NH3 - 0,0392 г Н2SO4
X = 0,0179 л NH3
3. Концентрация аммиака в жидкости:
10 мл раствора NH3 в воде - 0,0179 л NH3
1000 мл раствора NH3 в воде - X л NH3
Х = 1,79 л NH3/ л раствора NH3 в воде
1,79 л NH3 / л воды = 1,79×18 / 22,4 ×1000 = 1,44×IO-3 кмоль NH3/кмоль воды =
= Хк.
Отчетная таблица Таблица 1.2
|
Концентрация аммиака |
Скорость газовой смеси в колонне W, м/с |
Расход воздуха Gв, м3/ч |
Кy,  |
||||||
|
Yн, кмоль NH3/ кмоль воздуха |
Yк, кмоль NH3/ кмоль воздуха |
Хк, кмоль NH3/ кмоль воды |
Yн*, кмоль NH3/ кмоль воздуха |
Yк*, кмоль NH3/ кмоль воздуха |
DYср, кмоль NH3/ кмоль воздуха |
||||
1. Схема установки.
2. Отчетная табл. 1.2.
3. Расчетная часть.
4. Фазовая диаграмма У - Х.
Контрольные вопросы
1. Абсорбция и ее применение.
2. Способы выражения состава фаз.
3. Материальный баланс процесса абсорбции.
4. Понятия о молекулярной и турбулентной диффузиях.
5. Основное уравнение массоотдачи и массопередачи.
6. Средняя движущая сила процесса абсорбции и способы её расчета.
7. Поверхность контакта фаз при пленочном режиме работы.
6. Изображение процесса на фазовой диаграмме.
Литература [4, с. 227-265].
Лабораторная работа 2
ИЗУЧЕНИЕ ПРОЦЕССА РЕКТИФИКАЦИИ БИНАРНОЙ СМЕСИ

Методические указания
Во многих химических производствах необходимо производить выделение чистых веществ из смесей жидкостей. Одним из наиболее распространенных методов разделения смесей является ректификация. Этот метод основан на различии в температурах кипения, а, следовательно, и в летучести компонентов смеси. Если летучесть компонентов различна, то состав пара над жидкостью отличается от состава жидкой смеси бóльшим содержанием низкокипящего (легколетучего) компонента, который будем называть сокращенно НК. Вследствие этого при противоточном контакте жидкой смеси с парами, который осуществляется при ректификации в результате массообмена, пары будут обогащаться НК, а жидкость - высококипящим (труднолетучим) компонентом - ВК [1].
В конечном итоге пары будут представлять собой более или менее чистый НК, а жидкость - ВК.
Процесс ректификации осуществляется преимущественно в тарельчатых или насадочных колонных аппаратах периодическим или непрерывным способом.
Схема ректификационной установки непрерывного действия для разделения бинарной смеси представлена на рис. 2.1.
На ректификацию поступает исходная жидкая смесь, содержание в которой НК составляет хf. В процессе ректификации смесь разделяется на 2 части: часть, обогащенную НК - дистиллат и часть, обогащенную ВК - кубовый остаток. Обозначим в долях НК состав дистиллата - хр, а кубового остатка - хw.
Исходная смесь подается в ту часть колонны где жидкость имеет состав хf. Стекая вниз по колонне, она взаимодействует с поднимающимся вверх паром, образующимся при кипении кубовой жидкости в кипятильнике 2. Пар имеет начальный состав, примерно равный составу кубового остатка хw, т.е. является обедненным НК. В результате массообмена происходит переход НК из жидкости в пар, а ВК - из пара в жидкость [2].
В точке ввода исходной смеси пар может иметь в пределе состав, равновесный с исходной смесью. Для более полного обогащения пара НК верхнюю часть колоны орошают жидкостью состава хр, которая получается в дефлегматоре 3 путем конденсации пара, выходящего из колонны. Эта часть конденсата называется флегмой. Другая часть конденсата пара выводится из дефлегматора в виде продукта разделения - дистиллата. Жидкость, отводимая из нижней части колонны, называется кубовым остатком. Таким образом, в ректификационной колонне осуществляется непрерывный процесс разделения подаваемой в колонну исходной бинарной смеси на дистиллат с высоким содержанием НК и кубовый остаток, обогащенный ВК. Если обозначить количество поступающей на ректификацию исходной смеси Gf, количество получаемого дистиллата Gp и количество кубового остатка Gw, то материальный баланс выразится равенством: Gf
= Gp + Gw
Если заданы количество и состав исходной смеси и составы продуктов разделения, то для определения количеств дистиллата и кубового остатка уравнение дополняют уравнением материального баланса для НК:
Gf хf
= Gp хp + Gw хw
Степень разделения - чистота получаемых при ректификации продуктов - определяется физико-химическими свойствами разделяемых веществ, размерами и конструкцией аппарата, условиями проведения процесса (гидродинамикой, давлением и пр.).
На рис. 2.2 в координатах состав пара (у) - состав жидкости (х) графически представлена типичная зависимость между равновесными (линия 1) и рабочими (линия 2 и 3) составами фаз для бинарной смеси. Уравнения линий рабочих концентраций для верхней и нижней частей колонны имеют различный вид:
для верхней части:
yв = R/(R+1)x + xp / (R+1)
для нижней части:
yн = (R+F)/(R+1)x - (F-1)xw / (R+1),
где F = Gf/Gp - удельное количество поступающей исходной смеси; R = GR/Gp
- флегмовое число, представляющее собой отношение количества флегмы, подаваемой на орошение колонны к количеству дистиллата.
В уравнениях рабочих линий составы взаимодействующих фаз выражаются только в мольных долях НК.
При расчетах процессов ректификации принимаются следующие допущения, мало искажающие фактические условия протекания процесса, но значительно упрощающие расчет:
1. Разделяемая смесь следует правилу Трутона, согласно которому отношение мольной теплоты испарения или конденсации к абсолютной температуре кипения для всех жидкостей является приближенно величиной постоянной. Для смеси, состоящей из двух компонентов: rсм / Tсм = r1 / T1
= r2 / T2 » const.
Отсюда следует, что при конденсации 1 кмоль ВК в колонне испаряется 1 кмоль НК, т.е. мольное количество паров, поднимающихся по колонне постоянно.
2. Принимается, что yp = xp, a yw = xw, т.е. пренебрегается укрепляющим действием дефлегматора в процессе конденсации в нем паров и пренебрегается исчерпывающим действием кипятильника.
3. Теплоты смешения компонентов разделяемой смеси равны нулю.
Кроме того, в расчетах исходят из того, что смесь, подлежащая разделению, поступает в колонну нагретой до температуры кипения на питающей тарелке. В этом случае точка пересечения рабочих линий низа и верха колонны лежит на ординате x = хf.
Следует отметить, что питание колонны исходной смесью может осуществляться в виде пара и в виде жидкости с температурой ниже температуры кипения смеси.
Из анализа уравнений рабочих линий низа и верха колонны с учетом сделанных допущений следует, что рабочая линия верхней части колонны пересекает диагональ в точке А (при этом x = хр), а нижней - в точке В (x = хw). Между собой эти линии пересекаются в точке С, где x = хf. По высоте точка С располагается между равновесной линией и диагональю и зависит от R, при Rmin точка С находится на равновесной линии, при Rmax - на диагонали.
По фазовой диаграмме (рис. 2.2) графическим построением может быть найдено число теоретических ступеней изменения концентраций или число "теоретических тарелок". За "теоретическую тарелку" принимается такая условная тарелка, на которой (см. рис. 2.2) пар состава у1, взаимодействуя с жидкостью состава х1, изменяет концентрацию до равновесной y1*.
В этом случае число "теоретических тарелок" определяется построением ступеней между рабочими и равновесными линиями в пределах от хw до хр.
На реальных тарелках в колонне не достигается равновесное состояние между паром и жидкостью. Поэтому для аналогичного изменения концентрации требуется большее число действительных тарелок.
Отношение числа "теоретических тарелок" nт к числу действительных тарелок nд в колонне называют средним коэффициентом полезного действия тарелок (КПД колонны): hср = nт /nд
Величина КПД как мера интенсивности работы тарелок зависит от гидродинамических условий на тарелке, физических свойств пара и жидкости, кривизны равновесной линии. В связи с тем, что все эти факторы могут изменяться с изменением концентрации, КПД каждой тарелки h будет отличаться от среднего для всех тарелок hср. Величина hср определяется опытным путем.
Проведение npoцecca ректификации требует затраты тепловой энергии. Тепло, необходимое для испарения кубовой жидкости, подводится в кипятильник. Обычно подвод тепла осуществляется насыщенным водяным паром, а для колонн малого диаметра можно использовать электронагрев.
Теоретический расход тепла на проведение процесса определяется из уравнения теплового баланса: Qк = Gp
(R+1)rp + Gp cp tp + Gw
cw tw - Gf cf tf,
где rp - удельная теплота конденсации паров в дефлегматоре;
cf, cp, cw -
соответственно, теплоемкости исходной смеси, дистиллата и кубового остатка; tf, tp, tw - температуры кипения исходной смеси, дистиллата и кубовой жидкости, соoтвeтcтвeннo.

Описание лабораторной установки.
Технологическая схема экспериментальной установки показана на рис. 2.3. Она представляет собой часть ректификационной установки непрерывного действия для разделения бинарной смеси, а именно ее укрепляющую часть. Разделяемая смесь этилового спирта и воды заливается в куб колонны 1, снабженный мерным стеклом и выполняющий функции кипятильника. В нижней части куба установлены три электронагревателя. Образующиеся в кубе пары направляются в колонну 2. Давление паров замеряется дифманометром 3, установленным на приборной панели.
Рис. 2.3. Схема экспериментальной установки.
Ректификационная колонна с колпачковыми тарелками имеет диаметр 77 мм и высоту 950 мм. Колонна выполнена из отдельных царг высотой 190 мм, между которыми установлены колпачковые тарелки. Общее число тарелок в колонне 6. На каждой тарелке располагается по два колпачка. Площадь свободного сечения тарелки 4,5 × 10 -4
м2.
Пар, обогащенный этиловым спиртом, из колонны поступает в дефлегматор 4, в змеевик которого поступает охлаждающая вода. На поверхности змеевика пар конденсируется и поступает в флегмоделитель 5. Корпус флегмоделителя выполнен из стеклянного цилиндра, закрытого сверху и снизу резиновыми пробками. Сверху расположен штуцер для ввода жидкости из дефлегматора, снизу - два штуцера для ее отвода: правый штуцер служит для отвода дистиллата, левый - для возврата флегмы.
Разделение потоков осуществляется переключением поворачиваемой воронки, управляемой электромагнитным механизмом, связанным с реле времени. Когда магнит отключен, то под действием стальной шайбы поворачиваемая воронка находится в положении, изображенном сплошными линиями. При этом конденсат из дефлегматора подается в левый штуцер, а из него через гидравлический затвор в верхнюю часть колонны на орошение. Включенный магнит притягивает стальную шайбу, и, следовательно, перемещает воронку в положение, изображенное штрих-пунктирными линиями. В этом случае конденсат выводится через правый штуцер, поступает в мерное устройство 6 и сбрасывается в куб колонны.
Концентрация получаемого дистиллата измеряется ареометром, установленным в колбе 7.
Порядок проведения работы
Пуск установки выполняется только после детального ознакомления с её устройством и работой, инструкцией по технике безопасности и в присутствии преподавателя или лаборанта.
1. Предварительно необходимо установить по мерному стеклу наличие и с помощью ареометра состав исходной смеси в кубе. Для этого в колбу отобрать из куба через нижний кран 8 жидкость в количестве 150-200 мл и ареометром измерить ее плотность. После проведенных измерений жидкость слить в куб через верхний кран 9. Данные по равновесию смеси этиловый спирт-вода представлены в табл. 2.2.
2. Включить пускатель, установленный на стене и подать напряжение на установку.
3. ЛАТРом 10 по амперметру 16 подать ток 4А на обогрев колонны. Включить тумблеры 12,13,14 и подать напряжение на электронагреватели куба. ЛАТРом 15 по амперметру 11 установить ток 8,5 А на регулируемый электронагреватель. Включить потенциометр 17. Амперметр 22 контролирует работу нагревателей куба, включаемых тумблерами 12,13.
4. Вентилем 18 установить расход охлаждающей воды в дефлегматоре таким образом, чтобы ее температура на выходе из дефлегматора составляла не более 35-40 °С (что соответствует показаниям ротаметра в интервале 40-60 делений).
5. По дифманометру 3 следить за началом кипения смеси в кубе. При достижении разности уровней 30 мм выключить тумблеры 12 и 13 и ЛАТРом 15 поддерживать кипение смеси в кубе так, чтобы давление паров составляло 50-60 мм столба жидкости в дифманометре.
6. При появлении из дефлегматора дистиллата, стекающего в флегмоделитель, включить тумблер 19, подающий напряжение на обогреватель флегмы. Контроль за подаваемым напряжением осуществляется вольтметром 20.
7. В течение всей работы визуально следить за отсутствием вскипания флегмы в колбе с ареометром и поддерживать примерное равенство температур флегмы, поступающей в колонну, и паров, поступающих в дефлегматор.
8. При достижении постоянства регистрируемых прибором температур необходимо установить на реле времени 20 заданное преподавателем флегмовое число и включить тумблер 21 подачи напряжения на управляющий электромагнит флегмоделителя.
9. По истечении 10 - 15 минут работы установки с флегмоделителем и при повторном достижении постоянства температур, регистрируемых прибором КСП-4, что будет свидетельствовать о выходе колонны на установившийся режим, произвести снятие опытных данных в соответствии с табл. 2.1.
10. Для измерения производительности колонны по дистиллату необходимо перекрыть вентиль бюретки 6 и по секундомеру замерить время t получения 20 мл дистиллата. По окончании измерения полученный дистиллат слить в куб.
11. После окончания работы выключить тумблеры 14,19,21, подающие напряжение на нагреватель куба, флегму и электромагнит флегмоделителя.
ЛАТРом 10 снять напряжение с нагревателя обогрева колонны. Выключить контрольно-измерительный прибор КСП-4.
12. Через 20 минут прекратить подачу воды в дефлегматор и выключить пускатель на стене.
При работе установки нужно помнить, что этиловый спирт ядовит, он обладает наркотическим действием. В смеси с воздухом этиловый спирт взрывоопасен. Температура вспышки 16,1 оС. При обнаружении загазованности около установки необходимо ее выключить и включить вентиляцию.
Результаты замеров сводят в отчетную табл. 2.1.
Отчетная таблица Таблица 2.1
|
Производительность по дистиллату |
Температура, оС |
Расход охлаждающей воды |
Концентрация |
||||||||||
|
Исходной смеси в кубе, tf |
Паров, поступающих в дефлегматор, tp |
Конденсата из дефлегматора во флегмоделитель, tф |
Флегмы в колонну, tф¢ |
Воды, поступающей в дефлегматор, tн |
Воды, выходящей из дефлегматора, tк |
Кубовая жидкость |
Дистиллат |
||||||
|
Время получения 20 мл, с |
кг/с |
Показания ротаметра |
Gв, кг/с |
 |
 |
 |
 |
||||||
1. По полученным значениям плотностей смеси вода-этиловый спирт в кубе rw и дистиллате rp определить по диаграмме рис. 2.4 содержание спирта в % масс.


Содержание спирта в % массовых
Содержание спирта в % объемных
Рис. 2.4. Плотность водно-спиртовых растворов при разных температурах.
Пересчитать концентрацию смеси из массовых долей
хw =

где Мс , Мв - соответственно, мольные массы спирта и воды, кг/кмоль.
Аналогично рассчитать мольную концентрацию дистиллата - хр.
2. В координатах y-x построить на основе табл. 2.2 равновесную зависимость для смеси этиловый спирт-вода. На этом графике по результатам опыта построить линию изменения рабочих концентраций.
Равновесные данные системы этанол-вода: Таблица 2.2
|
х |
1,9 |
9,66 |
16,61 |
26,08 |
39,65 |
50,79 |
57,32 |
67,63 |
74,72 |
89,43 |
|
y |
17 |
43,75 |
50,69 |
55,80 |
61,22 |
65,64 |
68,41 |
73,85 |
78,15 |
89,43 |
|
t |
95,5 |
86,7 |
84,1 |
82,3 |
80,7 |
79,8 |
79,3 |
78, б4 |
78,41 |
78,15 |
где х - состав жидкой фазы в мольных процентах; у - состав паровой фазы в мольных процентах; t - температура смеси, °С.
3. Графическим построением ступеней изменения концентрации в пределах от хw до хр определить число "теоретических тарелок".
4. Рассчитать средний КПД колонны.
5. Производительность колонны по дистиллату Gp [кг/с] рассчитать по зависимости:
Gp = 2 × 10-5 rp / t
6. Рассчитать скорость паров в колонне wf [м/с], отнесенную к полному ее сечению: wf = Gp (R+1) × 22,4 (T+tp ) / (F×Mp×T),
где F - площадь сечения колонны, м2, если внутренний диаметр колонны 77 мм; tp - температура паров дистиллата, определяемая по табл. 2.2 для хр; Мр - мольная масса дистиллата, равная Мр = Мсхр
+ Мв(1-хр), кг/кмоль.
7. Составить тепловой баланс дефлегматора:
Qв
= Qп
Gв
св (tк - tн ) = Gp (R+1) [rp
+ cp (tp - tф )],
где Gв - расход охлаждающей воды, кг/с; св -
теплоемкость воды кДж/(кг × oC); tн, tк - температура охлаждающей воды, поступающей в дефлегматор и уходящей из дефлегматора, оС; tф
- температура конденсата, поступающего из дефлегматора в флегмоделитель, оС; rp - теплота конденсации паров дистиллата, кДж/кг: rp =
– теплота конденсации этилового спирта, кДж/кг; rв – теплота конденсации водяного пара, кДж/кг; сp - теплоемкость дистиллата, кДж/(кг × oC): сp =
Физические параметры св, rc, rв, cс
находятся по справочным таблицам [3] при соответствующих температурах.
Содержание отчета
1. Схема установки.
2. Расчетная часть.
3. На графике в координатах y-х определить число ступеней изменения концентраций.
4. Отчетная таблица.
Контрольные вопросы
1. Что такое ректификация?
2. Принципиальная схема ректификационной установки.
3. Материальный баланс ректификационной колонны.
4. Основные допущения, применяемые при описании процесса ректификации.
5. Вывод уравнений рабочих линий.
6. Построение рабочих линий на диаграмме y-x .
7. Флегмовое число.
8. Минимальное и максимальное флегмовое число.
9. Оптимальное и действительное флегмовое число.
10. Тепловой баланс ректификационной колонны.
Литература [4, с. 265-292].
Лабораторная работа № 3
изучение АДСОРБЦИИ В АППАРАТЕ С НЕПОДВИЖНЫМ СЛОЕМ ЗЕРНИСТОГО АДСОРБЕНТА

Цель работы: ознакомление с работой адсорбционной установки периодического действия. Определение коэффициентов массопередачи и защитного действия, предельной величины адсорбции при поглощении водяного пара цеолитом марки NaX или CaX.
Описание лабораторной установки.
Рис. 3.1. Схема лабораторной установки
Работа проводится на установке (рис. 3.1), которая работает по следующей схеме. Воздушный поток поступает из компрессора 1 через ресивер 9, игольчатый вентиль 2 и ротаметр 3. Далее он делится на две части: одна направляется в увлажнительную камеру 4, имеющую нагревательный элемент для нагрева воды и контактный термометр для поддерживания определенной температуры в камере, другая часть направляется в обход увлажнительной камеры. После смешения обоих потоков измеряется с помощью психрометра 5 влагосодержание воздуха, поступающего через 3-х ходовой кран 6 в колонну, заполненную адсорбентом. Влагосодержание воздуха после колонны 7 измеряется психрометром 8. Осушенный воздух выбрасывается в атмосферу.
Порядок выполнения работы
Лабораторная работа проводится в следующей последовательности.
1. Ознакомиться с содержанием работы и установкой.
2. Включить нагревательный элемент увлажнительной камеры 4 тумблером.
3. Убедившись, что вода в увлажнительной камере нагрелась
до необходимой температуры, 3-х ходовым краном соединить систему с атмосферой, включить компрессор 1 и игольчатым вентилем 2 установить по ротаметру 3 заданный расход воздуха.
4. После установления необходимой влажности соединить систему с адсорбционной колонкой, т.е. быстро переключить 3-х ходовой кран на слой адсорбента. С этого момента начинать отсчет времени опыта.
5. Сначала, через каждые 30 с - 6 замеров, затем через каждые 60 с - 7 замеров, а потом через каждые 120 с все остальные замеры записывать в табл. 3.1. показания психрометров на выходе и на входе колонки.
6. Опыт проводить до тех пор, пока показания психрометра 8 на выходе из колонки не будут одинаковыми с показаниями психрометра 5.
7. Окончив опыт, выключить нагреватель увлажнителя, затем - компрессор.
8. Опытные данные свести в табл. 3.1.
Отчетная таблица Таблица 3.1
|
t |
До колонки |
После колонки |
Расчетные данные |
|||||||||
|
с |
tc, °С |
tм, °С |
хо, кг/кг |
tc, °С |
tм, °С |
х, кг/кг |
хо/х |
ln(хо/х) |
ln(хо/х)-1 |
ао, кг/м3 |
К, с/м |
КV, 1/c |
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
13 |
|
... |
Длительность работы слоя адсорбента в стадии параллельного движения фронта сорбции (при L > Lo) рассчитывается по уравнению Н.А. Шилова [1]:
t = K ×
L - to,
где t - время защитного действия слоя, с; К - коэффициент защитного действия, с/м, показывающий время защитного действия слоя длиной 1 м; L - длина слоя, м; to - потеря времени защитного действия, с (кинетическая константа, пропорциональная скорости массообмена, при бесконечно большой скорости становится равной нулю).
При изучении сорбционного процесса обычно замеряют выходную кривую сорбции - изменение концентрации поглощаемого вещества во времени за слоем адсорбента (рис. 3.2). Затем, пользуясь развернутым уравнением динамики сорбции t = K × L – K × W × (ln (xo/x) -1) / KV,

строят график в координатах (ln (xo/x – 1)) = f (t).
Рис. 3.2. Выходная кривая сорбции Рис. 3.3. Кривая сорбции
Отрезок, отсекаемый на оси абсцисс (рис. 3.3), равен K×L, а котангенс угла наклона прямой есть K×W/KV. Здесь в уравнении W - объемная скорость потока, отнесенная к полному сечению аппарата, м3/(м2×c);
K = ao / (W × xo¢),
где ao - предельная величина сорбции, кг сорбированного вещества на 1м3 слоя адсорбента; KV - коэффициент массопередачи, с-1. Объемная скорость парогазовой смеси W определяется по уравнению расхода: V = W × S.
Таким образом, необходимо:
1. По температурам сухого tc
и мокрого tм
термометров определить влагосодержание воздуха на входе xо и на выходе х адсорбционной колонны для каждого момента времени: x = 0,622 × j × Pc / (735 - j × Pc),
где j - относительная влажность воздуха (отн. ед.) вычисляется по формуле:
j = [Pм – 0,494 (tс – tм)] / Pc,
где Рм, Рс - давление насыщенного водяного пара в воздухе при температуре мокрого tм и сухого tc термометров, мм рт. ст.
Давление насыщенного водяного пара может быть рассчитано по уравнению, аналогичному уравнению Антуана, которое для водяного пара в диапазоне температур от 0 до 60 °С имеет вид: ln P = 18, 76 – 4081,18 / (236,77 + t)
или P = e18,76 - 4081,18 / (236,77 + t)
2. Построить по экспериментальным данным выходную кривую сорбции в координатах: x = f (t) (рис. 3.2).
3. Рассчитать и построить выходную кривую сорбции в координатах:
(ln(xo/x) – 1) = f (t) (рис. 3.3).
4. Вычислить коэффициент защитного действия по отрезку, отсекаемому прямой на оси абсцисс К,
с/м: К = t1 / L.
5. Вычислить предельную величину сорбции по уравнению: ao = K × V × xo¢/ S,
где S - площадь сечения колонки, м2; V - объемный расход газовой cмеcи, м3/с; xo¢ - влажность газовой смеси, кг воды на 1м3
воздуха на входе в слой адсорбента: xo¢ = xo × r; r - плотность воздуха при температуре газовой смеси (при tc), рассчитанная по уравнению Клапейрона: r = M × 273 / [22,4 × (273 + tc)],
где М -
мольная масса газа, кг/кмоль.
6. Вычислить коэффициент массопередачи КV
по котангенсу угла наклона прямой (рис. 3.3): ctg a = - K × W / KV; KV = - K × W / ctg a
Содержание отчета
1. Схема установки.
2. Таблица опытных данных.
3. Выходная кривая сорбции в координатах х - t.
4. Выходная кривая сорбции в координатах (ln(xo/x) – 1) = f(t).
Характеристика адсорбентов:
Цеолит – СаХ.
Размер пор - 10 Ao.
Размер гранул: диаметр - 2 мм, длина - 3 мм.
rнас = 735 кг/м3.
Температура регенерации: 400 ¸ 450 °С.
Цеолит – NаХ.
Размер пор - 10 Аo.
Размер гранул - 3 мм.
rнас = 700 кг/м3
Температура регенерации: 400 ¸ 450 oС.
Условия проведения опыта.
1. Длина слоя цеолита: L = 4 ¸ 6 см.
2. Влажность воздуха: xo
= 15 ¸ 20 × 10-3 кг влаги / кг воздуха.
3. Температура воздуха: tc = 20 ¸ 25 oС.
4. Разность показаний сухого и мокрого термометров на входе:
tc
– tм = 0,2 ¸ 0,3 °С.
5. Расход газовой смеси: V = I8 ¸ 22 л/мин.
Контрольные вопросы.
1. Механизм процесса адсорбции.
2. Равновесие при адсорбции. Изотермы адсорбции.
3. Требования к адсорбентам. Промышленные адсорбенты.
4. Тепловой эффект при адсорбции.
5. Уравнение Шилова, время защитного действия, потеря времени защитного действия, понятие "проскока".
6. Конструкции адсорберов.
Литература [4, с. 366-393].
Лабораторная paбoта № 4
ИЗУЧЕНИЕ КИНЕТИКИ КОНВЕКТИВНОЙ СУШКИ
кАПИЛЛЯрно-Пористых МаТЕРИАлов
Цель работы: практическое ознакомление с методикой экспериментального изучения кинетики конвективной сушки и теоретическим описанием кинетических закономерностей процесса; выработка навыков использования расчетных методик тепломассообменных процессов.
Методические указания
Удаление влаги из твердых влажных материалов путем ее испарения и отвода образовавшихся паров при конвективном теплоподводе широко применяется во многих отраслях промышленности.

Агрегатное состояние, теплофизические свойства высушиваемых материалов, теплофизические характеристики сушильного агента являются основными факторами, влияющими на кинетику процесса и существенно меняющими закономерности его протекания. На кинетических кривых U(t) и T(t) принято выделять несколько характерных участков (рис. 4.1 и 4.2), соответствующих различным физическим механизмам процесса удаления влаги: прогрев материала до температуры мокрого термометра; период постоянной скорости сушки; период падающей скорости сушки и др. [I].
Рис. 4.1. Кривая сушки Риc. 4.2. Термограмма сушки
В частных случаях некоторые стадии процесса могут отсутствовать или вырождаться до незначительных по длительности, по сравнению с общим временем сушки, периодов. В настоящее время теоретически невозможно предсказать кинетику, поэтому расчетные методики строят на основе экспериментальных данных.
Описание лабораторной установки
Экспериментальная часть работы выполняется на конвективной циркуляционной сушилке (рис. 4.3), состоящей из вентилятора 1, замкнутого воздуховода 2, образующего циркуляционный контур, в который для подогрева воздуха встроен электрокалорифер 3. Центральная царга 4 циркуляционного воздуховода 2 используется как рабочая сушильная камера, для визуальных наблюдений и размещения образцов в ней имеется застекленная дверца 5.
Необходимая температура воздуха поддерживается регулирующим потенциометром 6, а заданная скорость устанавливается заслонкой 7. Для измерения влагосодержания сушильного агента в воздуховоде 2 установлены два психрометра 8, 9. «Мокрый» термометр увлажняется дистиллированной водой.

Взвешивание производится без выключения вентилятора, т.е. в потоке воздуха и, следовательно, можно, не нарушая условия опыта, снять кривую изменения веса образца в течение всего эксперимента - кривую сушки.
Термограмма сушки Т(t) - температурная кривая, характеризующая средне-объемную температуру образца в течение всего эксперимента, снимается потенциометром 16, соединенным с термопарой 14, которая впаяна в иглу 15.
Рис. 4.3. Схема экспериментальной установки.
Порядок выполнения работы.
1. После ознакомления с описанием и схемой установки по разрешению преподавателя включается установка: а) включается вентилятор; б) устанавливается заслонкой 7 заданная скорость воздуха; в) включается регулирующий 6 и записывающий 16 потенциометры; г) включается электрокалорифер 3.
2. После прогрева установки до заданной температуры открывают дверцу 5 и образец 10 с иглой 15, в которой запаяна термопара 14, помещают на траверсу 11. Закрывают дверцу и отсчитывают по весам 12 массу абсолютно сухого образца. Результаты заносят в табл. 4.1.
3. Извлекают образец из сушилки и производят замеры всех величин, указанных в табл. 4.1.
4. Затем образец увлажняется (поместить образец в ванночку с водой и выдержать 10 - 15 минут).
5. В увлажненный образец точно по центру вводят иглу с термопарой (рис. 4.3) и помещают образец в сушильную камеру.
6. В процесса сушки регистрируется масса материала и его температура. Замеры производят с интервалом 1 мин - 10 замеров, а затем с интервалом 3 мин - до постоянной массы образца, данные измерений заносят в табл. 4.2.
Отчетная таблица Таблица 4.1
|
Измеряемая величина |
Размерность |
Значение |
|
Масса абсолютно сухого материала, Gc Длина L, ширина b, толщина h образца Плотность образца, rм Размеры сушильной камеры, B ´ H Скорость воздуха, w Температура сухого термометра, tc Температура мокрого термометра, tм Барометрическое давление, П |
г м кг/м3 м м/с oC oС Па |
Отчетная таблица Таблица 4.2
|
№ |
Время от начала процесса |
Показания весов Gвл.м., г |
Количество влаги Gвл., г |
Влагосодержание |
Температура материала Tм, °С |
Скорость сушки d  % / с |
|||
Из эксперимента |
Расчетное |
||||||||
|
t, мин |
t, с |
||||||||
|
… |
1. По данным опыта для каждого замера рассчитывается влажность материала в массовых процентах:
- количество влаги в материале Gвл. = Gвл.м. - Gc,
- влажность материала

2. По полученным данным построить кривую сушки

3. Построить термограмму сушки Tм = f(t).
4. Выделить на кривой сушки

= f(t) характерные участки: прогрев материала; период постоянной скорости сушки; период падающей скорости сушки.
В том случае, когда можно принять, что сушка протекает в первом периоде, для дальнейшей обработки принять методику, изложенную в разделе А, если влагосодержание образца во втором периоде изменяется значительно, то для дальнейшей обработки использовать методику, изложенную в разделе Б.
Раздел А. 1. Определить скорость сушки в первом периоде, вычислив тангенс угла наклона кривой сушки к оси абсцисс:
N = dW/(F×dt) = A × tg a = A × a/b,
где N - скорость сушки, кг влаги /(м2 × с); t - время сушки, с; W - масса испаренной влаги, кг; F - поверхность фазового контакта, м2; А -
коэффициент пропорциональности, учитывающий переход от размерности
Раздел Б. 1. Дифференцированием кривой сушки получить скорость сушки
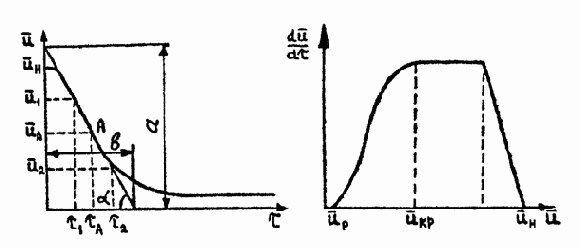
d
Рис. 4.4. Кривая сушки. Рис. 4.5. Скорость сушки.
Например, для момента времени tcр (точка А на кривой - рис. 4.4):
d


По этим данным построить кривую скорости сушки (рис. 4.5).
2. По показаниям «сухого» и «мокрого» термометров рассчитать парциальное давление водяных паров в сушилке Рс
и Рм и относительную влажность воздуха в сушилке j: j = Pc/Ps
× 100,
где Рs - давление насыщенного водяного пара в воздухе при температуре «сухого» термометра (ищется по таблицам свойств насыщенного водяного пара по tс), Па; Pc - парциальное давление водяного пара в сушилке при температуре «сухого» термометра, Па:
Рс = Рм
– В(tс
- tм) × П,
где Рм - давление насыщенного водяного пара при температуре «мокрого» термометра (ищется по таблицам свойств насыщенного водяного пара по tм), Па; П - барометрическое давление; В – коэффициент, зависящий от ряда факторов, из которых основным является скорость воздуха, для скорости воздуха w > 5 м/с:
В = 0,00001(65 + 6,75/w); (tс - tм) - разность температур «сухого» и «мокрого» термометров.
3. Вычислить равновесную влажность материала в %:

где j - относительная влажность воздуха подставляется в долях от единицы.
4. Рассчитать концентрации водяных паров у поверхности материала См и в ядре потока Сс, кг влаги/м3 воздуха:
См = Рм / [3,46 (273 + tм)]; Сс = Рс / [3,46 (273 + tс)],
где Рм и Рс
– подставляются в мм рт. ст.
5. При средней температуре пограничного слоя tср
= (tс + tм)/2 вычислить коэффициент массоотдачи: Nuд = E × Ren × Prд0,33 × Gu0,135,
где Nuд = b×L/D - массообменный критерий Нуссельта; Re = w×L×r/m - критерий Рейнольдса; Prд = m/(r×D) - диффузионный критерий Прандтля; Gu = (Тс-Тм)/Тс
- критерий Гухмана.
Здесь b - коэффициент массоотдачи, м/с; D -
коэффициент диффузии водяного пара в воздухе, м2/с; m - вязкость воздуха, Па×с; r - плотность воздуха, кг/м3; w - скорость воздуха, м/с; L - длина образца, м.
Коэффициенты Е и n выбираются в зависимости от значения критерия Рейнольдса из табл. 4.3:
Таблица 4.3
|
Re |
E |
n |
|
1 ¸ 2×102 |
0,9 |
0,5 |
|
2×102 ¸ 6×103 |
0,87 |
0,54 |
|
6×103 ¸ 7×104 |
0,347 |
0,65 |
Физические характеристики воздуха: Таблица 4.4
|
t, oC |
55 |
60 |
65 |
70 |
75 |
80 |
|
m×I06, Па×c |
20,01 |
20,25 |
20,48 |
20,71 |
20,94 |
21,17 |
|
r, кг/м3 |
1,076 |
1,060 |
1,044 |
1,029 |
1,014 |
1,000 |
|
D×I06, м2/c |
28,84 |
29,50 |
30,17 |
30,34 |
31,52 |
32,20 |
N = 200 × b (Cм - Сс) / (rм × h),
где N - скорость сушки в первом периоде, %/с; rм - плотность сухого материала, кг/м3; h - толщина материала, м.
7. По построенной кривой сушки (рис. 4.5) найти критическое влагосодержание

8. Рассчитать время первого периоде во формуле: t1 = (

где
9. Вычислить коэффициент скорости сушки: K = N / (
10. В интервале to < t < t1 рассчитать 5-7 значений влагосодержания материала в первом периоде:
11. Для ряда значений t ³ tкр рассчитать изменение влагосодержания во втором периоде (7-10 точек):
причем
12. Нанести полученные расчетным путем точки на график
Содержание отчета
1. Схема установки.
2. Отчетная таблица.
3. Расчет.
4. Графики
Контрольные вопросы
1. Методика проведения работы.
2. Массопередача в системах с твердой фазой.
3. Кинетические кривые.
4. Равновесное влагосодержание.
5. Формы связи влаги с материалом.
6. Расчет времени сушки.
7. Физический смысл коэффициента массоотдачи.
6. Свойства влажного воздуха.
9. Диаграмма I-x Рамзина.
10. Классификация сушилок.
Литература [4, с. 393-462].
Литература
1. Касаткин А.Г. Основные процессы и аппараты химической технологии. – М.: Химия, 1971. – 784 с.
2. Павлов К.Ф., Романков П.Г., Носков А.А. Примеры и задачи по курсу ПАХТ. – Л.: Химия, 1987. – 575 с.
3. Романков П.Г., Фролов В.Ф., Флисюк О.М., Курочкина М.И. Методы расчета процессов и аппаратов химической технологии (примеры и задачи). - Л.: Химия, 1993. - 496 с.
4. Плановский А.Н., Николаев П.И. Процессы и аппараты химической и нефтехимической технологии. – М.: Химия, 1987. – 496 с.
5. Процессы и аппараты пищевых производств: Лабораторный практикум / Под общей редакцией В.Н. Стабникова. – Киев: Вища шк., 1986. – 175 с.
6. Массопередача в системах с жидкой фазой: Лабор. работы. / Сост. Е.И. Преображенский, В.А. Набатов, В.Н. Куприянов, В.Н. Ладыжский. - Тамбовский ин-т хим. маш. - Тамбов, 1983. – 16 с.
7. Массообменные процессы с твердой фазой: Лабор. работы. / Сост. Т.В. Павлова, В.Б. Коробов, В.М. Нечаев, В.Н. Ладыжский. - Тамбовский ин-т хим. маш. - Тамбов, 1981. – 20 с.
8. Массообменные процессы: Лабор. раб. / Сост. Павлова Т.В., Набатов В.А., Куприянов В.Н., Коробов В.Б., Нечаев В.М. - Тамбовск. ин-т хим. машиностр. – Тамбов, 1991. – 48 с.
Учебное издание
ПРОЦЕССЫ И АППАРАТЫ ПИЩЕВЫХ ПРОИЗВОДСТВ
(массообменные процессы)
Лабораторные работы
для студентов заочного отделения специальности 1706
С о с т а в и т е л и:
Набатов Вячеслав Александрович;
Павлова Тамара Васильевна;
Нечаев Василий Михайлович;
Мозжухин Андрей Борисович;
Сергеева Елена Анатольевна.